ę°ë§ë´íźě¸íŹë ę°ë§ę¸°ě§ě íěěí¤ęł íŹëŞ
ěąě ě ě§íë¤[1,2]. ę°ë§ë´íźě¸íŹë ëěŹě ěźëĄ íěąě ě´ëŠ°, ę°ë§ë´íźě¸íŹě 죟ě ę¸°ëĽ ě¤ íëë íí뼟 íľí´ ę°ë§ę¸°ě§ěě ě´ě¨ęłź ě 체(fluid)뼟 ę°ë§ę¸°ě§ ë°ěźëĄ ë°°ěśěí¤ë ę˛ě´ë¤[1]. ëí¸ëĽ¨-욟뼨 ěë°ë
¸ě í¸ëŚŹíŹě¤ííě (Na+-K+ ATPase)ë ę°ë§ë´íźě¸íŹě 기ě 츥 ë§ě ěěší늰, ę°ë§ë´íźě íí 기ëĽě 기ěŹíë¤[3]. ę°ë§ë´íźě¸íŹë Na+-K+ ATPase ííëĄ ě¸í´ ë¤ëě ATP뼟 ěëšíë¤[3]. ATPë ěźë°ě ěźëĄ 미í ě˝ë댏ěě ë´ëś ë§(inner membrane)ěě ěěąëë¤[4]. ęą´ę°í 미í ě˝ë댏ěě 기ëĽě ę°ë§ë´íźě¸íŹě íí 기ëĽě ě¤ěíë¤[5]. ěšźě ęłźëśí, ě°í ě¤í¸ë ě¤ ë° ěľęˇźě ě¸ě§ë ěíŹě˛´(endoplasmic reticulum, ER) ě¤í¸ë ě¤ë ë
¸í, íšě¤ę°ë§ë´íźě´ěěŚ(Fuchsâ endothelial dystrophy), ěíŹę°ë§ëłěŚ(bullous keratopathy) ëą ę°ë§ë´íź ě§íęłź ě´ěíěě 체ě íě (ultrasound phacoemulsification)ëĄ ě¸í ę°ë§ë´íź ěěěě ëł´ě´ë ëłëŚŹíě ëłí뼟 íšě§ě§ëë¤[2,6,7].
ë
¸í ęłźě ě ę°ë§ě íŹëŞ
ěąęłź ęą´ę°ě ěíĽě ě¤ ě ěë¤[8]. ë
¸íë ę°ë§ë´íźě¸íŹ ěě¤ě ě¤ěí ěě¸ ě¤ íëě´ë¤[8]. ë
¸íë ę°ë§ëśě˘
ěźëĄ ě¸í´ ę°ë§ ě´ěě´ íěí ě¤ěí ěě¸ě´ë¤[9]. ë°ëźě ë
¸íë ę°ë§ë´íź ě§íě ěšëŁëĽź ěí 죟ě íě ěź ě ěë¤. ë
¸íë ě¸íŹë íŹę¸°ę° ë íŹęł , ë°ëźě ë
¸íëě§ ěě ě¸íŹě ëší´ ë í° ë¤ëŠ´ěą(polymegathism)ě ëł´ě¸ë¤[6,10,11]. ëí ë
¸íěěë ěąěĽ ě ě§ě ě 체 ě¸íŹ ěě ę°ěę° íšě§ě ě´ë¤[12]. ë°°ěë ě ě ěŹě ěě¸íŹěě ě¸íŹ ěě°ěŹë ë
¸íę° ë ěëĄ ë ëšë˛íë¤[12]. ě°í ě¤í¸ë ě¤ë ë
¸í뼟 ě ë°íë ę˛ěźëĄ ě ěë ¤ě ¸ ěë¤[13]. 미í ě˝ë댏ě/ě°í ě¤í¸ë ě¤ ę˛˝ëĄë ë
¸íě ę´ë ¨ëë¤[14].
Cyclosporine A (CsA)ë ěę° ëŠ´ě ě§íě ěšëŁ ë° ëě˘
ě´ěí¸ě 늴ěíě ęą°ëś ěë°Šě ěźë°ě ěźëĄ ěŹěŠëë 늴ě ěľě ě ě´ë¤[15]. CsAë ě´ěí¸ě ě쥴 ë° ę°ë§ ě´ě ěěŹěě ěśě ě§ě íĽěěí¤ë ę˛ěźëĄ ëł´ęł ëěë¤[15]. ěšźě/ěšźěë´ëŚ° ě í¸ ě ëŹ ę˛˝ëĄ(calcium-calcineurin signaling pathway)ë í¨ęłźě ě¸ ëŠ´ě ë°ěě íěíë¤[16]. CsAë ě¸íŹě§ě ěŹě´í´ëĄí댰(cyclophilin)ě 결íŠíěŹ ěšźěë´ëŚ°ě ěľě íë¤; ěšźěë´ëŚ°ě ěľě ë 늴ě ě¸íŹ ěŚěě ěľě íë¤[17]. CsAë ę°ë§ ě´ěëżë§ ěëëź ěęľŹęą´ěĄ°ěŚ ë° ěë 뼴기결ë§ěźě íŹí¨í ě꾏 í늴 ěźěŚě ěŹěŠëęł ěë¤[18,19]. ěęľŹęą´ěĄ°ěŚ íěěě 0.05% CsA ě ěě ěěě ěźëĄ ę°ë§ë´íźě ëłí뼟 ěźěźí¤ě§ ěë ę˛ěźëĄ ëł´ęł ë ë° ěěźë, ě´ ě°ęľŹěěë ěŹë ę°ë§ë´íźě¸íŹě ě¸íŹ ë°ëě íííě ěíĽë§ 쥰ěŹíë¤[20]. ëí ě¤ě§ ëŽě ëëě CsAë§ ëł´ęł ëěë¤. ě§ę¸ęšě§ ěŹë ę°ë§ë´íźě¸íŹě ë
¸íěě CsAě ěíĽě ëí ě°ęľŹë ě´ëŁ¨ě´ě§ě§ ěěěźëŻëĄ ěŹë ę°ë§ë´íźě¸íŹěě CsAě ěíĽě ëí ě°ęľŹę° íěíë¤. ě´ë˛ ě°ęľŹěěë ë°°ěë ěŹë ę°ë§ë´íźě¸íŹě ë
¸íě ëí CsAě í¨ęłźě ëí´ ěěëł´ęł ě íěë¤.
ëěęłź ë°Šë˛
ěŹë ę°ë§ë´íźě¸íŹě ëśëŚŹ ë° ë°°ě
본 ě°ęľŹë í댟ëíęľ ę°ë¨ěąěŹëłě ěěěíěŹěŹěěí(Institutional Review Board, IRB)/ě¤ëŚŹěěíěě ę˛í ë° ěšě¸íěë¤(ěšě¸ ë˛í¸: 2019-12-008). ě´ ě°ęľŹë íŹěąí¤ě ě¸ě ë°ëź ěíëěë¤. ě¸íŹë ě´ě ě 기ě ë ë°Šë˛ěźëĄ ë°°ěëěë¤ [15, 21]. 3-6ě°¨ëĄ ęłë ë°°ěě ęą°ěš ě¸íŹę° ěŹěŠëěë¤[2]. ě¸ ëŞ
ě ęłľěŹěëĄëśí° ěťě ę°ë§ě´ ěŹěŠëěë¤. ë°°ěë ěŹë ę°ë§ë´íźě¸íŹëĽź 0, 1, 10, 100 ÎźMě CsAě í¨ęť 2ěę° ëě ë°°ěíěë¤. ę°ë§ě Lions Eye Bank (Portland, OR, USA)ěě 꾏ě
íěěźëŠ°, 모ë ěĄ°ě§ ěíě ëí´ ěŹě ëě뼟 꾏íěë¤[21].
ë ¸í ë˛ í ę°ë˝í ěë¤ě (β-Galactosidase) ěźě
Senescence-associated β-galactosidase (SA-β-gal) ěźě í¤í¸(BioVision, San Francisco, CA, USA)뼟 ěŹěŠíěŹ ě¸íŹě ë
¸í뼟 íę°íěë¤[22]. ěě˝í늴, ě¸íŹëĄëśí° ěąěĽ ë°°ě§ëĽź ě ęą°í í ě¸íŹëĽź phosphate-buffered saline (PBS)ëĄ ě¸ě˛íěë¤. ě¤ě¨ěě ě¸íŹëĽź 15 ëś ëě ęł ě ěŠěĄě ęł ě ěěź°ë¤[22]. ě¸íŹëĽź PBSëĄ ě¸ě˛íęł , SA-β-gal ěźě ěŠěĄě ěŹěŠíěŹ 37°Cěě ë°¤ě ë°°ěíěë¤.
WST-8 í¤í¸ëĽź ěŹěŠí 미í ě˝ë댏ě íěěí¨ě(mitochondrial dehydrogenase)ě íěą ëśě
ě¸íŹëĽź 96-well plateěě 2ěę° ëě 0-100 ÎźM CsAëĄ ě˛ëŚŹíěë¤. 미í ě˝ë댏ě íěěí¨ěě íěąě WST-8 í¤í¸(Dojindo, Osaka, Japan)ëĄ ě¸Ąě ëěěźëŠ°, microplate reader (Spectramax Plus 384, Molecular Devices, San Jose, CA USA)뼟 ěŹěŠíěŹ 450 nměě íĄę´ë뼟 츥ě íěë¤[2]. 미í ě˝ë댏ě íěěí¨ěě íěąě ë쥰꾰(100%)ě ë°ąëśě¨(íęˇ Âą íě¤ í¸ě°¨)ëĄ ííëěë¤[2].
미í ě˝ë댏ě ë§ ě ě(ÎΨm)ě íę°
미í ě˝ë댏ě ë§ ě ě(ÎΨm)ë ë§ě´íŹëĄíë ě´í¸ ëśě(microplate assay)ěě JC-1 ěźëŁ(BioVision, San Francisco, CA, USA)뼟 ěŹěŠíěŹ ě¸Ąě ëěë¤[23]. Phenol red-free ë°°ě§ěě Black 96-well platesě seedingë ě¸íŹëĽź 2ěę° ëě 37°Cěě 0-100 ÎźMě CsAëĄ ě˛ëŚŹíěë¤. JC-1ě ěŹěŠíěŹ ěŹë ę°ë§ë´íźě¸íŹěě ÎΨmě ëłí뼟 ě
ěŚíěë¤. ę° ëëě CsAě ëí´ 4ę°ě wellě ěŹěŠíěë¤. Corresponding blank뼟 ě ě¸í í íę´ě ěë ę°ë뼟 ęłě°íěë¤[24]. JC-1ě ě ě/ë
šě íę´ ę°ëě ëšëĽź ęłě°íěë¤[23].
ě¸íŹ ë´ě 미í ě˝ë댏ěěěě reactive oxygen species (ROS) ěěą íę°
ě¸íŹ ë´ě 미í ě˝ë댏ěěěě ROS ěěąě ě°¸ęł ëŹ¸íě ë°ëź microplate assayěě dichloro-dihydro-fluorescein diacetate (DCFH-DA; Invitrogen, Carlsbad, CA, USA)ě MitoSOX (Invitrogen)뼟 ěŹěŠíěŹ íę°ëěë¤[2]. CsAě ę° ëëě ëí´ 6ę°ě wellě ěŹěŠíěë¤. DCF íę´ě ěë ę°ë뼟 ęłě°íěë¤[24]. 5 ÎźMě ěľě˘
ëëěě MitoSOXę° wellě 첨ę°ëěë¤.
미í ě˝ë댏ě ë° ě¸íŹě§ě ěšźě ěěš íę°
미í ě˝ë댏ě ë° ě¸íŹě§ě ěšźě ěěšë microplate assayěě ę°ę° Rhod-2 AM (Invitrogen)ě Fluo-4 AM (Invitrogen)ě ěŹěŠíěŹ ě¸Ąě ëěë¤[25]. Rhod-2 AM (1 ÎźM) ëë Fluo-4 (1 ÎźM)ę° ěśę°ëěěźëŠ°, ꡸ í ě´ëě´ ęłłěě 20ëś ëě 37°CëĄ ë°°ěíěë¤. íę´ ę°ë뼟 츥ě í기 ěí´ ëśę´ęł(spectrofluorometer)뼟 ěŹěŠíěë¤. ę° ëëě CsAě 4ę°ě wellě´ ěŹěŠëěë¤. íę´ě ěë ę°ë뼟 ęłě°íěë¤[23,24].
ě¸íŹ 죟기 ëśě
NucleoCounterÂŽ (NC-3000â˘, ChemoMetec, France)ě ěí ě¸íŹěŁźę¸° ëśěě ë¤ěęłź ę°ě´ ěíëěë¤[26]. ěŚ, ě¸íŹëĽź 1 Ă 106 cells ë°ëëĄ íë ě´í
íęł 2ěę° ëě ë°°ěíěë¤. 24ěę° í, ëśě ě¸íŹ ë° ě ě°Šěą ě¸íŹę° ěťě´ěĄë¤. ě¸íŹëĽź ě°¨ę°ě´ 70% ěíěŹëĄ ęł ě ěí¤ęł , 0.1% Triton X-100, RNase A (30 Îźg/mL), 0.1% ethylenediaminetetraacetic acid ë° íëĄíźë ěě´ě¤ë¤ě´ë(propidium iodide) (50 Îźg/mL) (pH 7.4)ě í¨ě í ěźě ěŠěĄęłź í¨ęť ë°°ěíěë¤[26]. NucleoCounterÂŽ (NC-3000â˘)뼟 ěŹěŠíěŹ ě¸íŹ DNA í¨ëě ëśěíë¤[26]. ě¸íŹ 죟기 ëśíŹ ëśěěë ěľě 10,000ę°ě ě¸íŹę° ěŹěŠëěë¤[26].
ě¨ě¤í´ ë¸ëĄŻ(Western blot)ě ěí ë¨ë°ąě§ ë°í ëśě
ě¨ě¤í´ ë¸ëĄŻě íě¤ ę¸°ë˛ě ěŹěŠíěŹ ěíëěë¤[21]. ę°ëľí 기ě íě늴, ě¸íŹ ë¨ë°ąě§ě phosphatase inhibitor cocktail (PhosSTOP; Roche, Basel, Switzerland)ě protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA)ě íŹí¨í radioimmunoprecipitation assay buffer (Biosesang, Seoul, Korea)뼟 ěŹěŠíěŹ ěśěśëěë¤[21]. 15ëś ëě 16,000 Ă gěě ěěŹ ëśëŚŹí í ě츾ěĄě glycogen synthase kinase (GSK) 3β, phosphorylated glycogen synthase kinase (pGSK) 3β, extracellular signal-regulated kinase-1/2 (ERK1/2) ë° phosphorylated extracellular signal-regulated kinase-1/2 (pERK1/2)ě 츥ě ě ěŹěŠíěë¤. ě¸íŹ ë¨ë°ąě§ě sodium dodecyl sulfate polyacrylamide gel electrophoresis뼟 ě¤ěíęł , polyvinylidene difluoride ë§(Immobilon-P; Millipore Corp., Bedford, MA, USA)ěźëĄ ěŽę¸°ęł 5% íě§ě ěě 30ëś ëě ë°°ěíěë¤. ꡸ ë¤ě anti-human mouse ERK1/2 antibody (Abcam, Cambridge, MA, USA), anti-human pERK1/2 rabbit antibody (Abcam), anti-human GSK3β rabbit antibody (Abcam) ëë anti-human pGSK3β rabbit antibody (Abcam)ě ę°ě ěźě°¨ í체ě í¨ęť ë°°ěíěë¤. 5-bromo-4-chloro-3-indolylphosphate/nitro blue tetrazolium ë°ě 기ě§(Promega, Madison, WI, USA)ě ě´ě°¨ě ě욟댏-ě¸ě°ëśí´í¨ě(alkaline-phosphatase) ě íŠ í체ě í¨ęť 3ěę° ëě ë°°ěë í 늴ě ë°ěěą ë¨ë°ąě§ ë°´ëě ę˛ěśě ěŹěŠëěë¤. ë°ě´í°ëĽź ě ëíí기 ěí´ ě´ëŻ¸ě§ ëśě ěě¤í
(Chemidoc XRS, Bio-Rad, Hercules, CA, USA)ě ěŹěŠíë¤[21].
Zoula occludens 1 (ZO-1)ě 늴ě íę´ ěźě
ěŹë ę°ë§ë´íźě¸íŹëĽź 24-well ë°°ě íë ě´í¸ě seedingíęł PBSěě 3.7% íŹëŚěë°íë ěŠěĄě 15ëś ëě ęł ě ěěź°ë¤[2]. ě¤ě¨ěě 5% ěźě íě˛ęłź í¨ęť 30ëś ëě ë°°ěí ě¸íŹëĽź 2ěę° ëě anti-human ZO-1 rabbit antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA)ě í¨ęť ë°°ěíěë¤. ě´ě´ě, í루ě¤ë ě¸ě¸ ě´ěí°ě¤ěěě°ěź(fluorescein isothiocyanate)ě´ ě íŠë anti-rabbit goat IgG antibody (1:100)ě í¨ęť 2ěę° ëě ě´ë ěěě ě¤ě¨ ë°°ěíěë¤. íľ ěźěě Hoechst 33342 dye (1:1000 íŹě; Invitrogen)ëĄ ěíëěë¤[21].
결 곟
ěŹë ę°ë§ë´íźě¸íŹě ěëł ë° ë°°ěë ěŹë ę°ë§ë´íźě¸íŹě ë ¸í
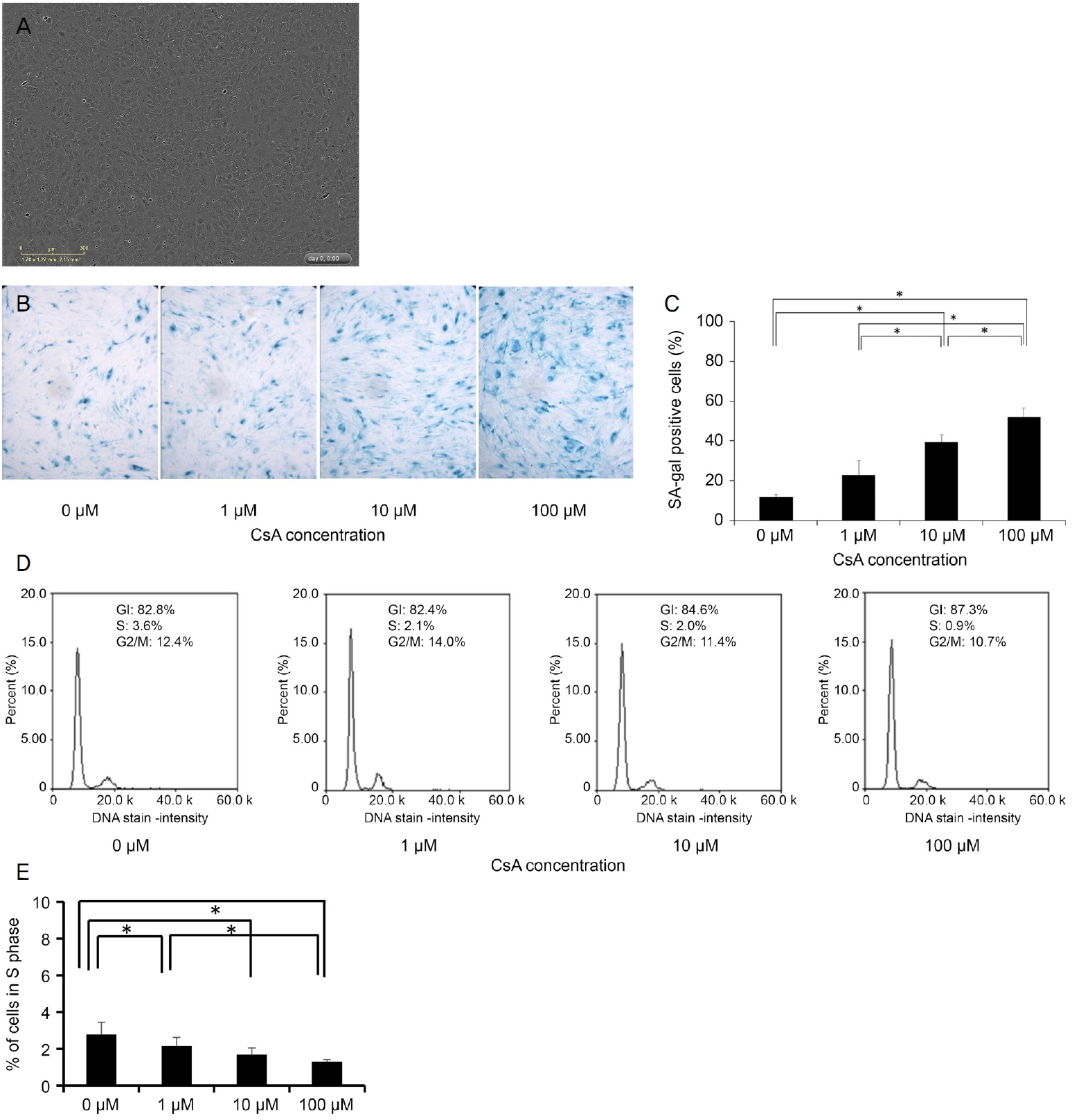
P0ěě ë°°ěë 1ě°¨ ę°ë§ë´íźě¸íŹë ě체 ë´ěě ëł´ě´ë ę˛ęłź ě ěŹí ěĄę°í íí뼟 ëł´ěë¤. ě¸íŹë 모ěě´íŹ í¨í´ě ëł´ěŹěŁźěë¤(Fig. 1A) [21]. SA-β-gal ěźěě ěí´ ë
¸í뼟 츥ě íěë¤. SA-β-gal-ěěą ě¸íŹě ë°ąëśě¨ě CsAě ëëě ë°ëź ěŚę°ěěź°ë¤(p=0.003, Kruskal-Wallis test) (Table 1, Fig. 1B, C). SA-β-gal-ěěą ě¸íŹě ë°ąëśě¨ě ë쥰꾰곟 ëšęľíěŹ 10, 100 ÎźMěě ëěë¤(p=0.029, Man-Whitney U test). ě¸íŹěŁźę¸° ëśěě CsAę° S기ě ě¸íŹ ë°ąëśě¨ě ę°ěěěź°ěě ëł´ěŹěŁźěë¤(p=0.007) (Table 1, Fig. 1D, E). CsAë ë쥰꾰ě ëší´ 1, 10 ë° 100 ÎźMěě S기ě ě¸íŹ ë°ąëśě¨ě ę°ěěěź°ë¤(p=0.029, Mann-Whitney U test). S기 ě¸íŹě ë°ąëśě¨ě 100 ÎźMěě 1 ÎźMě ëší´ ë ëŽěë¤(p=0.029).
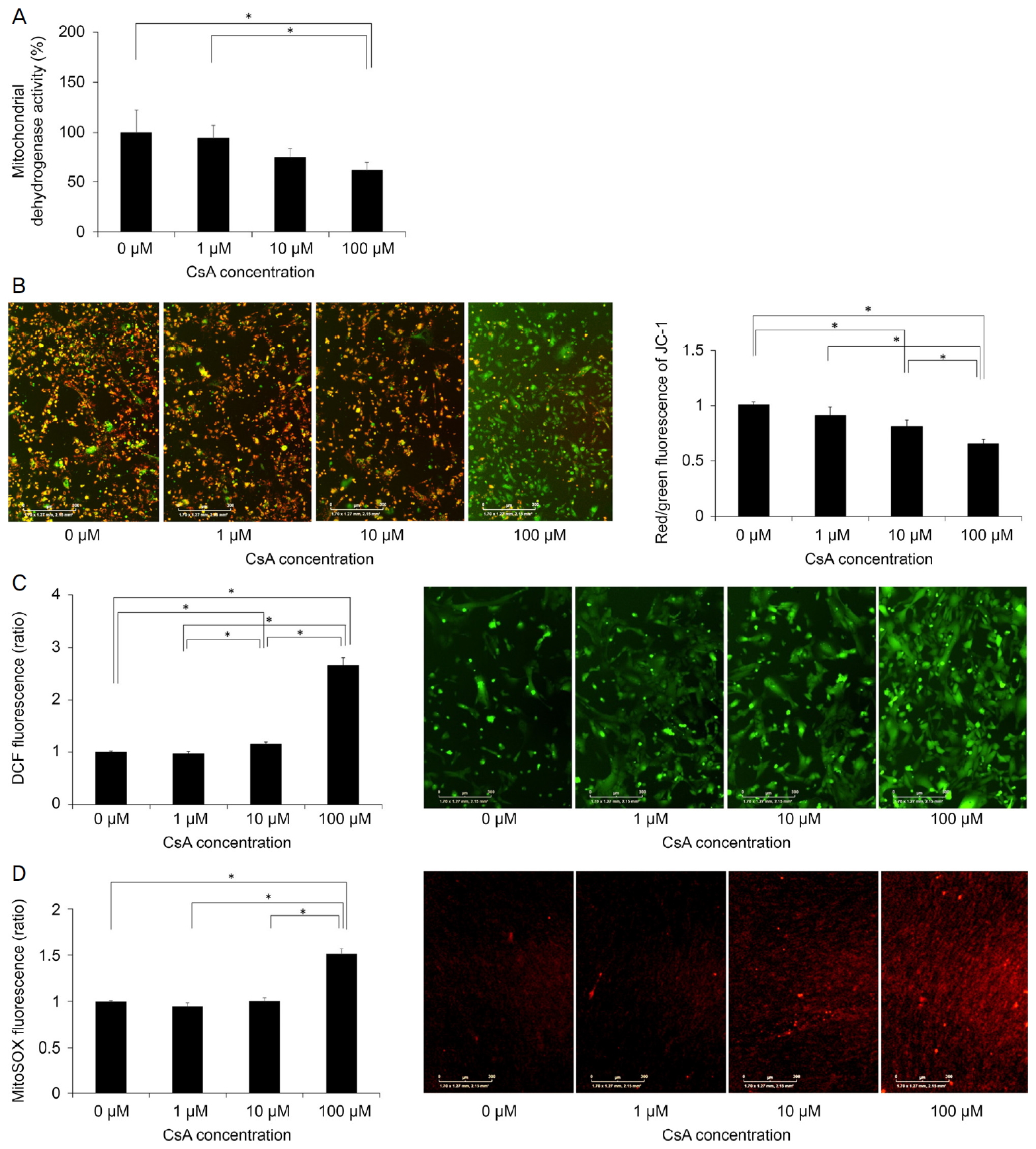
미í ě˝ë댏ě íěěí¨ě íěą ë° ÎΨmě ëí CsAě í¨ęłź
WST-8 ëśěě ěí´ íę°ë 미í ě˝ë댏ě íěěí¨ěě íěąëśěěě, CsAë ë쥰꾰곟 ëšęľíěŹ 100 ÎźMěě 미í ě˝ë댏ě íěěí¨ěě íěąě ę°ěěěź°ë¤(p=0.029, Mann-Whitney U test) (Table 1, Fig. 2A). JC-1ě ěŹěŠíěŹ ÎΨmě 츥ě íěë¤. CsAë ëëę° ěŚę°í¨ě ë°ëź ÎΨmě ę°ěěěź°ë¤(p=0.004, Kruskal Wallis test) (Table 1, Fig. 2B). ÎΨmě ë쥰꾰곟 ëšęľíěŹ 10 ÎźM ë° 100 ÎźMěě ë ëŽěë¤(p=0.029, Man-Whitney U test). ÎΨmě ë쥰꾰, 1 ÎźM ë° 10 ÎźM ëëě CsAě ëšęľíěŹ 100 ÎźMěě ë ëŽěë¤(p=0.029).
ě¸íŹ ë´ě 미í ě˝ë댏ěěě ROSě ëí CsAě í¨ęłź
ě¸íŹ ë´ ROS íěąě DCF íę´ě ěí´ ě¸Ąě ëěë¤[24]. ě¸íŹ ë´ ROSë CsAě ëëě ë°ëź ěŚę°íěë¤(p=0.005, Kruskal Wallis test) (Table 1, Fig. 2C). CsAë 10 ÎźMěě ë쥰꾰곟 1 ÎźMě ëšíěŹ, 100 ÎźMěěë ë쥰꾰, 1 ÎźM, 10 ÎźMě ëšíěŹ ě¸íŹ ë´ ROS ěěšëĽź ěěšěěź°ë¤(p=0.029). 미í ě˝ë댏ě ROS íěąě MitoSOX red íę´ě ěí´ ě¸Ąě ëěë¤. CsAë ë쥰꾰, 1 ÎźM, 10 ÎźMęłź ëšęľíěŹ 100 ÎźMěě 미í ě˝ë댏ěě ROS ěěšëĽź ěěšěěź°ë¤(p=0.029) (Table 1, Fig. 2D).
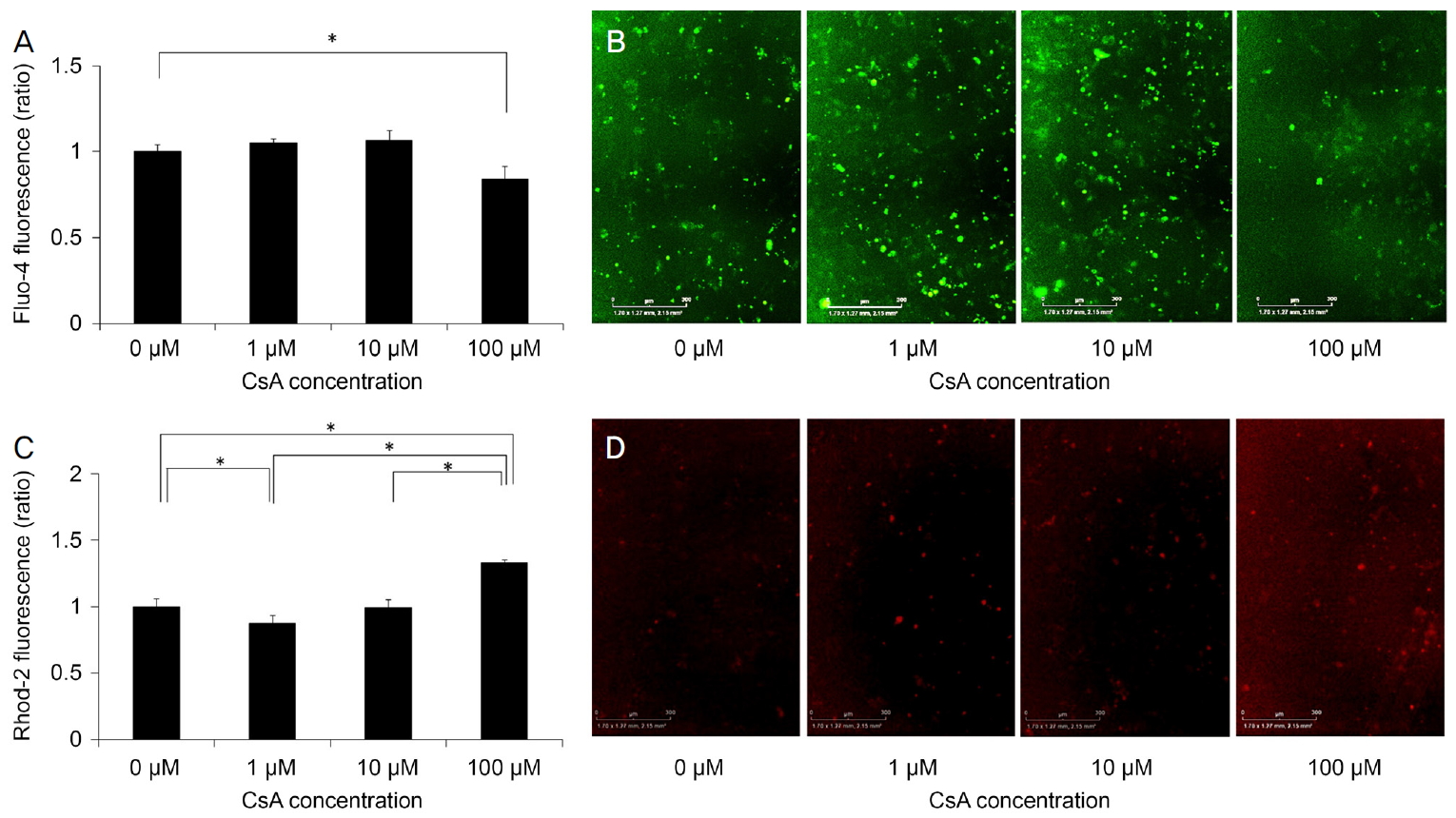
ě¸íŹ ë´ ë° ëŻ¸í ě˝ë댏ěě ěšźěě ëí CsAě ěíĽ
ě¸íŹ ë´ ěšźěě Fluo-4ě ěí´ ě¸Ąě ëěë¤[25]. CsAë 100 ÎźMěě ëěĄ°ęľ°ëł´ë¤ ěšźě ěěšëĽź ę°ěěěź°ë¤(p=0.029) (Table 1, Fig. 3A, B). 미í ě˝ë댏ěě ěšźě ěěšë Rhod-2ě ěí´ ě¸Ąě ëěë¤[25]. Rhod-2 íę´ ę°ëë CsAě ëëę° ëěě§ě ë°ëź ěŚę°íěë¤(p=0.008, Kruskal-Wallis test) (Table 1, Fig. 3C, D). CsAë ë쥰꾰곟 ëšęľíěŹ 1 ÎźMěě 미í ě˝ë댏ě ěšźě ěěšëĽź ę°ěěěź°ęł 100 ÎźMěěë ěěšěěź°ë¤(p=0.022, p=0.001).
늴ě íę´ ěźěě ë°ëĽ¸ ZO-1 ë°í
ZO-1 (ë
šě)ě ëí´ ëŠ´ě íę´ ěźěě ěííěë¤. CsAë ëë ě쥴 ë°ŠěěźëĄ ZO-1 ë°íě ę°ěěěź°ë¤(Fig. 4D).
ęł ě°°
ěźě í ěě ě¸íŹëśě´ í, ě ě ě¸íŹ ěŚěě ëšę°ěě ěźëĄ ě ě§ëęł , ě´ íěě ë
¸íëźęł íë¤[13]. ë
¸í ě¸íŹë ëěŹě ěźëĄ ęłě 기ëĽě íě§ë§, ě íë ě¸íŹ ëśě´, ę°ěë ěąěĽ ěë, íííęł íëë ě¸íŹ íí ë° ě¸íŹěŁźę¸° 쥰ě ë¨ë°ąě§ě ëłí뼟 ëł´ě¸ë¤[27]. ě´ ě°ęľŹěě, CsAë ë°°ěë ěŹë ę°ë§ë´íźě¸íŹěě ë
¸í뼟 ě ëíěë¤. CsAëĄ ě˛ëŚŹí í ë
¸í ě¸íŹě ěë ëë ě쥴ě ěźëĄ ěŚę°íěë¤. ě¸íŹěŁźę¸° ëśěě S기ě ě¸íŹ ěę° ě ě´ CsAę° ě¸íŹěŁźę¸° ě ě§ëĽź ě ëí¨ě ëł´ěŹěŁźěë¤. S기 ě ě§ěěë G0/G1 ë¨ęłěě ěśě ëë ë
¸í ě¸íŹěě ë§ë¨ ě¸íŹěŁźę¸° ě ě§ëĽź ëł´ě¸ë¤[28].
CsAě ě꾏 ěźěŚ ě§íě ęľěě ěšëŁ ě¸ěë ęą´ě , ëĽë§í°ě¤ěą ę´ě ěźęłź ę°ě ěę°ëŠ´ě ě§íě ěšëŁë ę°, 콊íĽ, ěŹěĽ ëąě ěĽę¸° ě´ě í 늴ěěľě 뼟 ěí´ ěŹěŠëęł ěë¤[29]. 기쥴ě ëŞëŞ ě°ęľŹěěë CsAëĄ ě¸í ë§ěą ě ëśě ęłź ę°ě ě ë
ěąęłź ě¸íŹ ë
¸íě ě ëę° ëł´ęł ëěë¤[30-32]. Justo et al [33]ě ë°°ěë 콊íĽě¸ę´ ěíźě¸íŹěě 10 Îźg/mL, 8.3 mM CsAě ěí´ ěŹëŹ ë¨ęłě apoptotic pathwayę° ě ëë¨ě íě¸íěěźëŠ°, 죟ěí apoptotic pathwayëĄ mitochondrial injury뼟 ëł´ęł íěë¤. Mandavilli et al [34]ě 미í ě˝ë댏ě DNAě ěěě´ íëłľëě§ ěěźëŠ´, ëłíë í¨ěę° electron transport chaině ëě
ë¨ěźëĄě¨ ěě ëźëěšźëĄ ě¸í ěěě ę°ěíěí¤ęł ęśęˇšě ěźëĄ ë ë§ě 미í ě˝ë댏ě DNA ëě°ëłě´ ë° oxidant productioně ě´ëíë¤ęł íěë¤. ę˛°ęľ ëŻ¸í ě˝ë댏ě ROSě ěě°ęłź DNA ëě°ëłě´ě ě
ěíě ě¸íŹě ë
¸íëĄ ě´ě´ě§ ě ěë¤[34].
ě§ę¸ęšě§ ě ě ě§íě ěšëŁěě CsAě ëłľěŠěźëĄ ě¸í ę°ë§ë´íźě¸íŹě ë
¸íě ëí ě°ęľŹë ěě§ ëł´ęł ë ë° ěë¤. 본 ě°ęľŹë ë°°ěë ěŹë ę°ë§ë´íźě¸íŹěě CsAě ěí ë
¸íě í¨ęłźě ëí´ ëśěíěë¤. ě´ ě°ęľŹěě ě°ëŚŹë CsAě ěŹë ę°ë§ë´íźě¸íŹ ë°°ě í 미í ě˝ë댏ěě ëłíě ě´ě ě ë§ěśěëë° ëŻ¸í ě˝ë댏ěë ě¸íŹ ë´ ROSě 죟ě ęłľę¸ěě´ëŠ° ë
¸í ęłźě ěě íěě ě¸ ěí ě íë¤[35]. ë
¸í ě¤ě 미í ě˝ë댏ě 기ëĽě´ ě¤ë¨ë늰, 미í ě˝ë댏ě 기ëĽě ÎΨmě ę°ě ë° ęłźě°í돟 ěěąě ěŚę°ě ěí´ íę°ë ě ěë¤[36].
ÎΨmě ATP ěěąě´ëźë 미í ě˝ë댏ěě ě댏 ę¸°ëĽ ě ě§ě ě¤ěíë¤[37]. íŹęłźěą ě ě´ ě¸ęłľ ę°ë°Š(permeability transition pore opening)ě 미í ě˝ë댏ě 기ëĽě ëšę°ěě ěźëĄ ě í´í ěíĽě¸ ę˛ěźëĄ ëł´ęł ëěë¤[36,38]. íŹęłźěą ě ě´ ě¸ęłľ ę°ë°Šě íě ÎΨmě ěě¤, 매í¸ëŚě¤ ě´ě¨ě ë°Šěś ë° ě¸íŹě§ëĄě 미í ě˝ë댏ě ëěŹě ěě¤ě íŹí¨íë¤[38]. ÎΨmě íě í ěě¤ě ěëě§ëĽź ęł ę°ěí¤ęł 결곟ě ěźëĄ ě¸íŹ ěŹëŠ¸ě ě´ëíë¤[36]. ęł ěŠëě ě°í ě¤í¸ë ě¤ë ÎΨmě íě ěě¤ě ě ë°í ě ěë¤[39]. 본 ě°ęľŹěě, ë°°ěë ę°ë§ë´íźě¸íŹěě ęł ëëě CsAë ëë ě쥴ě ë°ŠěěźëĄ ÎΨmě ę°ěěěź°ë¤.
본 ě°ęľŹěě CsAę° ě¸íŹ ë´ ROS ěěšëĽź ěŚę°ěí¤ë ę˛ěźëĄ ë°íěĄë¤. ě°íě ě¤í¸ë ě¤ë p53 ě ěŹ ë°ěě íěąí, ěí´ëŚ°-ěěĄ´ěą í¤ëě (cyclin-dependent kinases) ěľě ě ě 쥰ě ë° í
ëĄëŻ¸ě´ ë¨ěśě íľí´ ë
¸í뼟 ě ëíěŹ ęśęˇšě ěźëĄ DNA ěě ë°ěě ě ë°íë¤[13]. ě´ ě°ęľŹěě, 미í ě˝ë댏ě ě°í ě¤í¸ë ě¤ë 100 ÎźM CsA ě˛ëŚŹ í ěěší ę˛ěźëĄ ëł´ěë¤.
ě¸íŹ ë´ ěšźě ěěšë 100 ÎźM CsA ě˛ëŚŹěě ę°ěíěě§ë§, 미í ě˝ë댏ě ěšźě ěěšë 100 ÎźM CsA ě˛ëŚŹ í ěěšíěë¤. CsAë 미í ě˝ë댏ěëĄëśí°ě ě˝ëźę˛ ëšë-ě ëë ěšźě ë°Šěśě ě§ě°ěí¤ë ę˛ěźëĄ ěë ¤ě ¸ ěë¤[6]. CsAë ě¸íŹ ë´ ěšźě ěěšëĽź ëíë´ë Fluo-3 íę´ě 2ë°° ěŚę°ëĽź ě ëíë¤[40]. 미í ě˝ë댏ěë ěšźěě´ě¨ě íěěą ěĄ°ě ě ě°¸ěŹíęł ě¸íŹě§ ěšźě ě í¸ëĽź 쥰ě í ě ěë¤[41,42]. ëěŹ ęłźě ě ěšźě ě쥴ě 쥰ě ě 미í ě˝ë댏ě 매í¸ëŚě¤(matrix)ěě ěźě´ëë¤[43].
GSK3ë ě¸íŹě ě¸íŹě§, 미í ě˝ë댏ě ë° íľěě ë°ę˛Źëë¤[44]. GSK3ě ę¸ëŚŹě˝ę˛ ëěŹěě ě¤ěí ěí ě íë ę¸ëŚŹě˝ę˛ íŠěąě (glycogen synthase)뼟 ě¸ě°í ë° ěĄ°ě íë¤[45]. íľęłź 미í ě˝ë댏ěě GSK3ë ě¸íŹě§ěě ëł´ë¤ ë íëě ě´ë¤[44]. GSK3βë ě ěĽ íí ëë ě ě°ěěŚěźëĄ ě¸í ě°í ě¤í¸ë ě¤ëĄ ě¸í 미í ě˝ë댏ě ę¸°ëĽ ěĽě ě ę´ë ¨ě´ ěë ę˛ěźëĄ ëł´ęł ëěë¤[46]. ERK1/2 경ëĄë ě¸íŹę° ě¸íŹ ě¸ ě í¸ëĽź ě¸íŹ ë´ ë°ěěźëĄ ě íěí¤ë ě í¸ ě ëŹ ëŞ¨ëě´ë¤[47]. ERK1/2 경ëĄë ě ě ě ë°í, íę´ íěą, ě¸íŹ ëśí, ěŚě ë° ę¸°ëĽě 쥰ě ě íŹí¨íë¤[47]. ERKě íěąíë ë
¸í ë°ěě ě ë°í기 ěí´ MKK3/6-p38 경ëĄě ěꡚěźëĄ ě´ě´ě§ë¤[48]. ERK ë§¤ę° ë
¸íë ě¸íŹěŁźę¸° ě§í, 미í ě˝ë댏ě ę¸°ëĽ ë° ě¸íŹ ě í¸ ě ëŹě íěí ë¨ë°ąě§ě íëĄí
ěě˘ ěěĄ´ěą ëśí´ëĽź íŹí¨íë¤[49]. ERK1, ERK2 ëë MEK ěľě ě ë ë
¸í ëŠěť¤ëěŚě íěąí뼟 ë°Šě§íë ę˛ěźëĄ ěë ¤ě ¸ ěë¤[49].
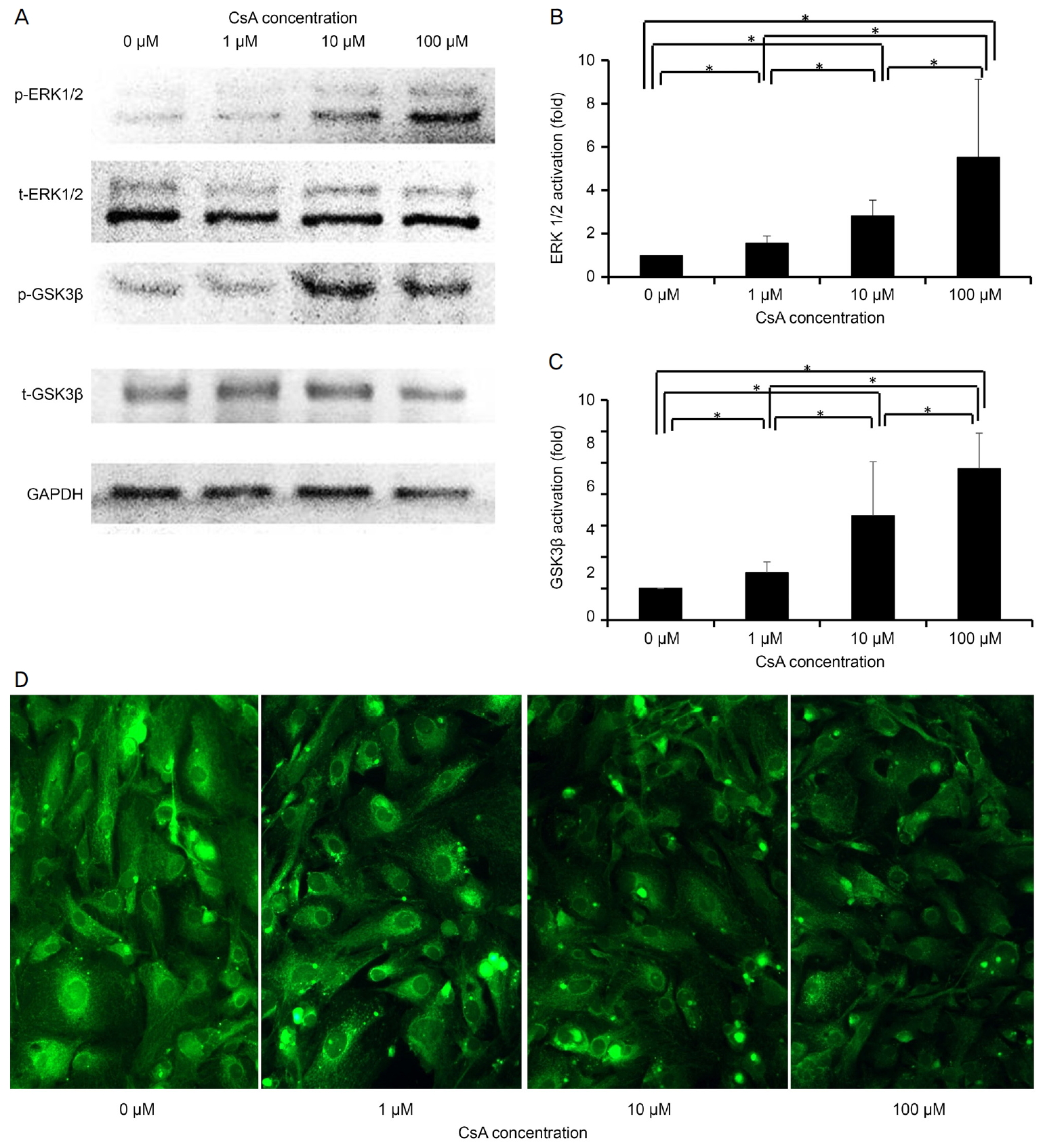
ě´ ě°ęľŹěě CsAë GSK3β ě í¸ ę˛˝ëĄ ë° ERK1/2 ě í¸ ę˛˝ëĄëĽź íěąíěěź°ëë°, ě´ë ě¸íŹě ë
¸í ë°ěęłź ë°ě í ę´ë ¨ě´ ěë¤. GSK3βě ę¸°ëĽ ëłíë ë
¸íě ěë°ëë íěěźëĄ, GSK3βë ěëě§ ěëšě ěě°ě ęˇ í 쥰ě ě ěě¤ě ě미íë¤[46]. ERK1/2 경ëĄě íěąíë íěą ě°ěě ěŚę°ě í¨ęť ë
¸í뼟 ě´ě§íë¤[48]. CsAë ěźë°ě ěźëĄ ěšźě/욟모ë댰(calcium/calmodulin) ěěĄ´ěą ě¸ëŚ°/í¸ë ě¤ë(serine/threonine) ë¨ë°ąě§ íŹě¤ííě ě¸ ěšźěë´ëŚ°(calcineurin)ě ěľě íë¤[50]. ěšźěë´ëŚ°ě ě¸ëŚ°-9ěě GSK3β뼟 íě¸ě°ííë¤[50].
본 ě°ęľŹěě CsAë ZO-1 ë°íě ę°ěěěź°ëë° ě´ë ëśě°Šěą ë° tight junction ě íŠ ë¨ë°ąě§ě´ë¤[51]. CsAë ZO-1ě ííĽ ěĄ°ě ě ě ëíë ę˛ěźëĄ ëł´ęł ëěë¤[46]. ZO-1 ë°íě ERK/p38 MAPK/JNK 경ëĄě íěąíě ěí´ ěĄ°ě ëë¤[52]. ę˛°ëĄ ě ěźëĄ, CsAë 미í ě˝ë댏ě-, GSK3β- ë° ERK1/2- ě쥴ě 경ëĄëĽź íľí´ ěŹë ę°ë§ë´íźě¸íŹěě ë
¸í뼟 ě ëíęł ZO-1 ë°íě ę°ěěí¨ë¤.
본 ě°ęľŹë ë°°ěë ěŹë ę°ë§ë´íźě¸íŹě ëí ëšě체 ě¤íěźëĄ ě체 ë´ě ě ěŠí기ěë ě´ë ě ë íęłę° ěě ę˛ěźëĄ ěę°ëë¤. ëí ę°ë§ě ě ěě ëĄ ěŹěŠëěě ę˛˝ě° ę°ë§ěíź, ę°ë§ę¸°ě§ě íľęłźíěŹ ę°ë§ë´íźě ëëŹíë ëëë ë§¤ě° ě ě ę˛ěźëĄ 기ëëë¤. ꡸ëŹë ęł ëëëĄ CsAę° ěŹěŠëë 경ě°ěë ę°ë§ë´íźěë ęł ëëëĄ ëëŹí ę°ëĽěąě´ ěě´ ě´ě ëí ëśěěŠě´ ě°ë ¤ëë¤. ë°ëźě ě체 ë´ ěŹë ę°ë§ë´íźě¸íŹě ë
¸íěë ěíĽě 미ěšëě§ě ëí ëśěě ěí´ ěśí ěě ěąě´ ëł´ěĽë ě체 ë´ ě°ęľŹę° íěíë¤.
íěŹ íŹëë§ěź ëąě ě꾏ěźěŚ ě§í ë° ěęľŹęą´ěĄ°ěŚ íěě ěšëŁěŠ ě ěěĄěźëĄ CsAę° ěŹěŠëęł ěë¤[53]. 본 ě°ęľŹ 결곟ě ë°ëĽ´ëŠ´ CsAě ëëě ë°ëź ěŹë ę°ë§ë´íźě¸íŹě ë
¸íę° ě ëëě´ ęł ëë CsAě ěí ěšëŁ í¨ęłźěë ě´ë ě ë ě íě´ ěě ę˛ěźëĄ ěę°ëë¤. ë°ëźě ě´ě ę°ě ëśěěŠě ě¤ě´ę¸° ěí´ ęł ëëě CsAę° íŹí¨ë ě ěěĄ ěŹěŠ ě , íěě ę°ë§ë´íźě¸íŹ íę°ę° ě´ëŁ¨ě´ě ¸ěź í ę˛ě´ë¤. ëí CsAě ěšëŁěě ěŹë ę°ë§ë´íźě¸íŹě ë
¸íë ěľěíěí¤ęł , ěšëŁ í¨ęłźëĽź ěľëëĄ ěťě ě ěë ë°Šë˛ě ëí ěëě ë¤ěí ě°ęľŹę° ě§íëě´ěź íë¤.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print